3. Identifier mapping with BridgeDb¶

3.2. Graphical Overview¶
This recipe will cover the highlighted topics
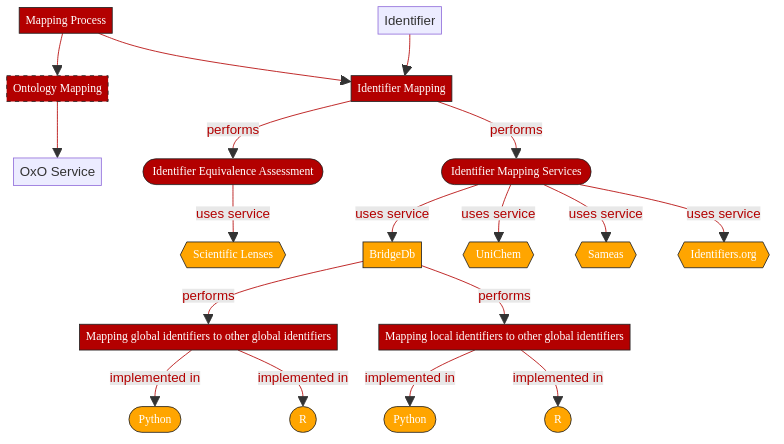
Fig. 3.1 Overview of key aspects in Identifier Mapping¶
3.3. Requirements¶
This recipe has the following requirements:
Recipe dependency:
Skill dependency:
Programming knowledge
R
Python
Technical dependency
R or Python environment
BridgeDb R package installed
3.4. Tools¶
The table below lists the software that is used to execute the examples in this recipe.
Software |
Description |
version |
Biotools record |
|---|---|---|---|
BridgeDb is a framework to map identifiers between various databases. It includes a Java library that provides an API to work with identifier-identifier mapping databases and resources. |
0.0.9 |
||
Python |
An interpreted, high-level and general-purpose programming language. |
3.8.5 |
|
pandas is a fast, powerful, flexible and easy to use open source data analysis and manipulation tool, built on top of the Python programming language. |
1.1.3 |
||
R |
R is a programming language and free software environment for statistical computing and graphics supported by the R Foundation for Statistical Computing. |
v4.0.3 |
|
tidyverse is an opinionated collection of R packages designed for data science. |
1.3.0 |
||
An R package for BridgeDb |
2.0.0 |
3.5. Identifier mapping with BridgeDb¶
Interlinking data from different sources is an essential step for data reusability and interoperability. This step requires dedicated tools. With the present recipe, we show how to use BridgeDb to carry out this process.
BridgeDb is an open source tool dedicated to performing identifier mapping 2. BridgeDb offers three different interfaces:
Java API
R package
REST Web-services
📖 In the context of this recipe, we distinguish between two types of identifiers:
Local identifiers which refer to identifiers that are minted within an organization or database and thus internally defined (i.e. local to said organization).
Global identifiers which refer to identifiers that are globally unique and uniquely point to an entity, as available from BridgeDb’s data sources file
We will focus here on two distinct cases, depending on the nature of the incoming data. Namely, whether our data is already using global identifiers or only relies on local identifiers.
In this recipe, we will cover how BridgeDb’s R package and webservices can be used to map between resource identifiers.
3.5.1. Mapping a global identifier to other global identifiers¶
In this case, the input data is a list of elements with an identifier that is part of BridgeDb’s data sources. In our example, we will use a list of Homo Sapien Hugo Gene Nomenclature Convention (HGNC) gene identifiers stored in a TSV file. The objective is to map these to other available gene identifiers.
3.5.1.1. BridgeDb via Webservices using Python¶
❗ For this tutorial Python v3.8.5, pandas v1.1.3, and BridgeDb Webservices v0.9.0 were used.
One of the biggest benefits of using BridgeDb webservices is that these can be accessed using most programming languages. Python has become one of the leading programming languages in data science and predictive modelling. Despite the lack of a dedicated BridgeDb Python library, we show here how to use the BridgeDb Webservices to perform exemplary mappings.
First, the required libraries need to be set up through several ‘pip install’ command (in the terminal)
## Install pandas:
pip install wheel
pip install pandas
## Install API calls library
pip install requests
Second, load the required libraries in your Python code:
##Load library to work with pandas data formats:
import pandas
##Check the version of this library:
pandas.__version__
##Load library to post get requests :
import requests
Then, we start by defining strings containing the URL of the webservices and the specific method from the Webservices we want to use. In our case, a batch cross reference. When doing the query, we need to specify the organism and the source dataset. We can also optionally specify a target data source if we only want to map to a specific data source, e.g. Ensembl.
url = "https://webservice.bridgedb.org/"
batch_request = url+"{org}/xrefsBatch/{source}{}"
If the aim is to map only to a specific target data source, then one can check whether the mapping is supported by invoking the following webservice call:
mapping_available = "{org}/isMappingSupported/{source}/{target}"
query = url+mapping_available.format(org='Homo sapiens', source='H', target='En')
requests.get(query).text
This will return True if the mapping between the given source and target is supported for the given organism or False otherwise.
We then load our data into a pandas dataframe and call the requests library using our query.
query = batch_request.format('?dataSource=En', org='Homo sapiens', source='H')
response = requests.post(query, data=data.to_csv(index=False, header=False))
The webservice response is now stored in the response variable. We can then simply pass this variable to the to_df method provided in the bridgedb_script.py module (see Code). This method will extract the response in text form and turn it into a pandas Dataframe with conveniently named columns and structured data.
The output table will contain the:
Original identifier
Data source that the identifier is part of
Mapped identifier
Data source for the mapped identifier
In our case the output of to_df is:
original |
source |
mapping |
target |
|---|---|---|---|
A1BG |
HGNC |
ENSG00000121410 |
En |
A1CF |
HGNC |
ENSG00000148584 |
En |
A2MP1 |
HGNC |
ENSG00000256069 |
En |
If we were to not specify the target data source (by passing an empty string as the parameter), we would get all the potential mappings for the given identifiers. In our case (top 10 rows):
original |
source |
mapping |
target |
|---|---|---|---|
A1BG |
HGNC |
uc002qsd.5 |
Uc |
A1BG |
HGNC |
8039748 |
X |
A1BG |
HGNC |
GO:0072562 |
T |
A1BG |
HGNC |
uc061drj.1 |
Uc |
A1BG |
HGNC |
ILMN_2055271 |
Il |
A1BG |
HGNC |
Hs.529161 |
U |
A1BG |
HGNC |
GO:0070062 |
T |
A1BG |
HGNC |
GO:0002576 |
T |
A1BG |
HGNC |
uc061drt.1 |
Uc |
A1BG |
HGNC |
51020_at |
X |
As one can see, using the BridgeDb webservice via Python is extremely simple and can be easily integrated in an annotation pipeline.
3.5.1.2. BridgeDb via the dedicated R package¶
After having loaded the required R libraries, we read the data and create a new column to include the source of the identifier.
data_df <- read_tsv(filepath, col_names=c('identifier'))
data_df$source = 'H'
We then load the data for the organism we are mapping from.
location <- getDatabase('Homo sapiens')
mapper <- loadDatabase(location)
And use the library’s dedicated function to map the identifiers:
mapping = maps(mapper, data_df, target='En')
This will return:
identifier |
source |
target |
mapping |
|---|---|---|---|
A1BG |
H |
En |
ENSG00000121410 |
A1CF |
H |
En |
ENSG00000148584 |
A2MP1 |
H |
En |
ENSG00000256069 |
As seen earlier when using Python language, we can obtain all possible mappings simply by not specifying the target. This will result in (top 10)
identifier |
source |
target |
mapping |
|---|---|---|---|
A1BG |
H |
Uc |
uc002qsd.5 |
A1BG |
H |
X |
8039748 |
A1BG |
H |
T |
GO:0072562 |
A1BG |
H |
Uc |
uc061drj.1 |
A1BG |
H |
Il |
ILMN_2055271 |
A1BG |
H |
U |
Hs.529161 |
A1BG |
H |
T |
GO:0070062 |
A1BG |
H |
T |
GO:0002576 |
A1BG |
H |
Uc |
uc061drt.1 |
A1BG |
H |
X |
51020_at |
Warning
An error message indicating “Error in download.file” may be thrown. This may be caused by the timeout variable being set to too small a value. To remediate the issue, try increasing the timeout variable value by calling options(timeout=300).
3.5.2. Mapping local identifier to a different global identifier¶
Note
In this section, we assume that we already have an equivalence file containing the mapping of a local identifier to one of the global identifiers. In our case, this will be contained in a TSV where we map our local gene identifier to HGNC. One may consult the list of other potential data formats in the Interlinking data from different sources recipe. The mapping should be one-to-one for this recipe.
The TSV mapping file looks as follows:
local |
source |
|---|---|
aa11 |
A1BG |
bb34 |
A1CF |
eg93 |
A2MP1 |
You may notice the source identifiers correspond with those used in the previous example.
This is how the mapping will work
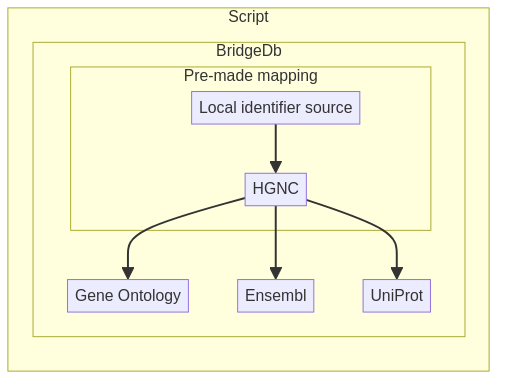
Fig. 3.2 Overview of BridgeDb tools¶
3.5.2.1. Webservices in Python¶
As before, we will define variables including the web-service's URL and the method that we will use, in this instance: xRefsBatch.
We then pass the source column to the post request as follows
source_data = case2.source.to_csv(index=False, header=False)
query = batch_request.format('', org=org, source=source)
response2 = requests.post(query, data = source_data)
You may notice here that we did not pass a target source, this could be done as specified before. Then, we use to_df again and as expected obtain the same dataframe as before.
To see the equivalences with our local identifiers, we can simply join the dataframes, as follows:
local_mapping = mappings.join(case2.set_index('source'), on='original')
which will return the following table (first 10 rows)
original |
source |
mapping |
target |
local |
|---|---|---|---|---|
A1BG |
HGNC |
uc002qsd.5 |
Uc |
aa11 |
A1BG |
HGNC |
8039748 |
X |
aa11 |
A1BG |
HGNC |
GO:0072562 |
T |
aa11 |
A1BG |
HGNC |
uc061drj.1 |
Uc |
aa11 |
A1BG |
HGNC |
ILMN_2055271 |
Il |
aa11 |
A1BG |
HGNC |
Hs.529161 |
U |
aa11 |
A1BG |
HGNC |
GO:0070062 |
T |
aa11 |
A1BG |
HGNC |
GO:0002576 |
T |
aa11 |
A1BG |
HGNC |
uc061drt.1 |
Uc |
aa11 |
A1BG |
HGNC |
51020_at |
X |
aa11 |
In case we did specify the target argument to be Ensembl (En), we would instead get
original |
source |
mapping |
target |
local |
|---|---|---|---|---|
A1BG |
HGNC |
ENSG00000121410 |
En |
aa11 |
A1CF |
HGNC |
ENSG00000148584 |
En |
bb34 |
A2MP1 |
HGNC |
ENSG00000256069 |
En |
eg93 |
Here, we see a one-to-one relation between the identifiers in HGNC and En while the relation between HGNC and UCSC Genome Browser (Uc) or Gene Ontology (T) is one-to-many. Depending on the identifiers and resources, the relation could also be many-to-many as shown below.
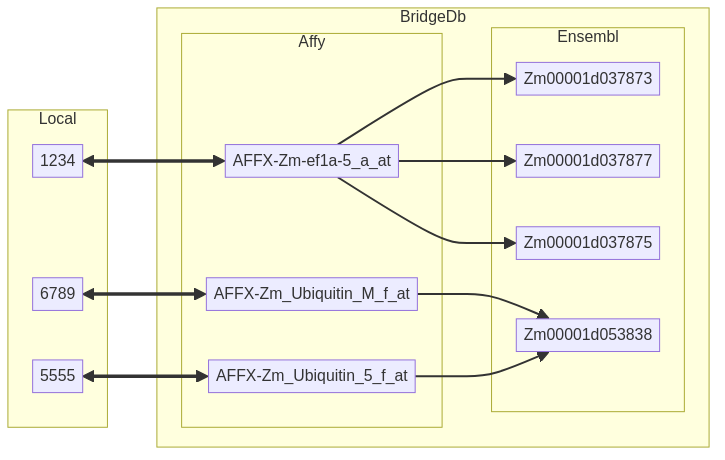
Fig. 3.3 An example of a mapping via BridgeDb. You may notice that despite the 1-to-1 relation between local and original we get a N-to-N relation between local and mapping due to the N-to-N relation between original and mapping.¶
Note
This many-to-many relationship stems from different scientific lenses in the data sources. You can read more about these in 1. The core idea is that depending on the domain/application of the data we can consider different entities as unique. While certain proteins could be considered “equal” from a biological perspective they may require differentiation when using a chemical len. This is what then leads to many-to-many relationships.
3.5.2.2. R Package¶
Here, we will follow the same steps as in the previous case. The only difference is that we need to specify the columns/fields to use when loading the data:
data_df <- read_tsv(filepath, col_names=c('local', 'identifier'))
Then, after computing the mapping, we can join it with the local identifier
right_join(data_df, mapping)
Assuming we did not specify the target data source we obtain the following table (first 10 rows):
local |
identifier |
source |
target |
mapping |
|---|---|---|---|---|
aa11 |
A1BG |
H |
Uc |
uc002qsd.5 |
aa11 |
A1BG |
H |
X |
8039748 |
aa11 |
A1BG |
H |
T |
GO:0072562 |
aa11 |
A1BG |
H |
Uc |
uc061drj.1 |
aa11 |
A1BG |
H |
Il |
ILMN_2055271 |
aa11 |
A1BG |
H |
U |
Hs.529161 |
aa11 |
A1BG |
H |
T |
GO:0070062 |
aa11 |
A1BG |
H |
T |
GO:0002576 |
aa11 |
A1BG |
H |
Uc |
uc061drt.1 |
aa11 |
A1BG |
H |
X |
51020_at |
In case we did specify the target data source we would get:
local |
identifier |
source |
target |
mapping |
|---|---|---|---|---|
aa11 |
A1BG |
HGNC |
En |
ENSG00000121410 |
bb34 |
A1CF |
HGNC |
En |
ENSG00000148584 |
eg93 |
A2MP1 |
HGNC |
En |
ENSG00000256069 |
3.6. Provenance¶
BridgeDb provides provenance information through:
A call to
/properties/method of the WebservicegetProperties()in BridgeDbR (passing the mapper as a parameter)
This returns the following information for each of the data sources for a given organism:
Data source name
Build date
Series
Data type
Data source version
Schema version
Improvements on provenance are under way (see here).
3.7. Code¶
You can find ready-made methods to map using R and Python for the given use cases here. These assume the data has the structure described in this recipe.
3.8. Conclusion¶
We showed how to use BridgeDb’s webservices and R package to map identifiers from different data sources using a minimal dataset. BridgeDb provides handy functionality to make ‘omics’ data more interoperable and reusable. As with all annotation services, it is important to bear in mind the version of the service being used as well as the data on which the service invokation has been performed. These are aspects of information provenance which we plan to provide in the future.
3.9. References¶
references
- 1
Colin Batchelor, Christian Y A Brenninkmeijer, Christine Chichester, Mark Davies, Daniela Digles, Ian Dunlop, Chris T Evelo, Anna Gaulton, Carole Goble, Alasdair J G Gray, Paul Groth, Lee Harland, Karen Karapetyan, Antonis Loizou, John P Overington, Steve Pettifer, Jon Steele, Robert Stevens, Valery Tkachenko, Andra Waagmeester, Antony Williams, and Egon L Willighagen. Scientific Lenses to Support Multiple Views over Linked Chemistry Data. In The Semantic Web – ISWC 2014. Lecture Notes in Computer Science, 16. 2014. doi:10.1007/978-3-319-11964-9_7.
- 2
Martijn P. van Iersel, Alexander R. Pico, Thomas Kelder, Jianjiong Gao, Isaac Ho, Kristina Hanspers, Bruce R. Conklin, and Chris T. Evelo. The BridgeDb framework: standardized access to gene, protein and metabolite identifier mapping services. BMC Bioinformatics, 11(1):5, January 2010. URL: https://doi.org/10.1186/1471-2105-11-5 (visited on 2021-02-05), doi:10.1186/1471-2105-11-5.
3.10. Authors¶
Authors
Name |
ORCID |
Affiliation |
Type |
ELIXIR Node |
Contribution |
|---|---|---|---|---|---|
Maastricht University |
|
Writing - Original Draft |
|||
University of Oxford |
|
Writing - Review & Editing |
|||
Heriot Watt University |
|
Writing - Review & Editing |