Plant genomic and genetic variation data submission to EMBL-EBI databases¶

Main Objective¶
Plant genotyping and phenotyping data are often used in combination to make evidence-based inferences about different trait expressions. A challenge here is not necessarily to collect the data, but to offer them in a stable way over the long term in public repositories with sufficient metadata information to make conclusions about them, in line with FAIR principles. Since these repositories are often not linked directly, it is even more important to provide metadata that allows users to recognise these links by their identifiers. A key point here is sample management: the identifiers assigned here help both humans and machines to understand which experimental data are linked.
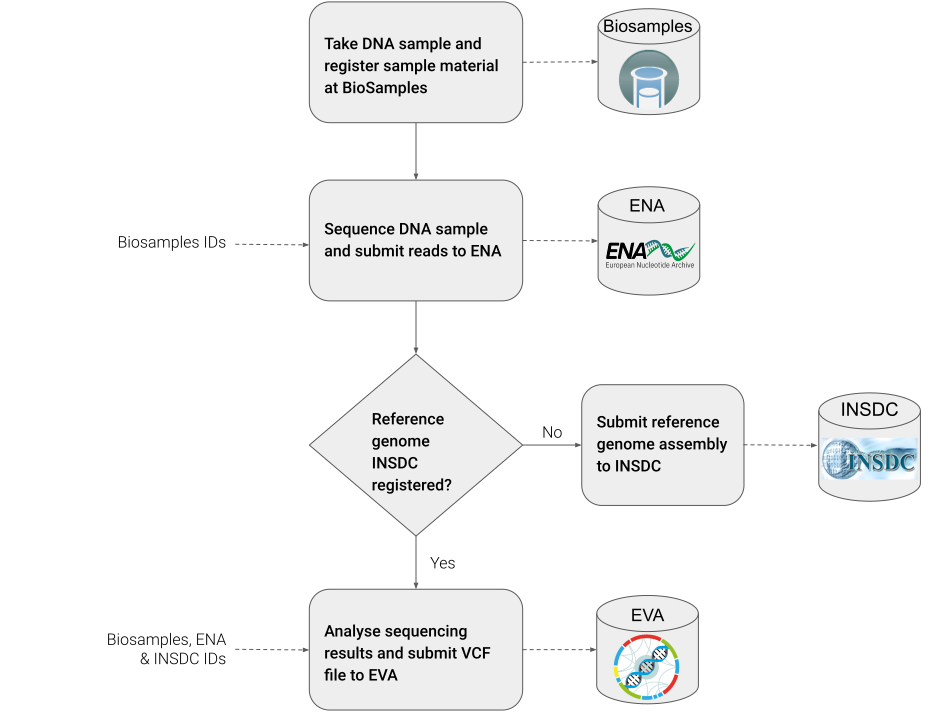
The main objective of the recipe is to provide a means of submitting to public repositories and tracking genotyping data, with a particular focus on plants. This includes:
1) Submission of sample data and metadata information to BioSamples.
2) Submission of sequencing data and metadata to ENA.
3) Retrieval of the correct genome assembly for the genotyping experiment
4) Conversion of the resulting analysis file (in VCF format) to be FAIR
5) Submission of the genotyping results to EVA.
In terms of FAIRification goals, this means obtaining stable, resolvable identifiers for the datasets and meeting community annotation requirements as expressed in the MIAPPE requirements.
Summary¶
This recipe provides guidance for submitting plant genotyping data to public repositories. It explains in a step-wise fashion which work should be done and when. Special attention should be paid to the metadata maintenance of the data that will be deposited in different repositories as part of this recipe. A prerequisite for fully understanding this recipe is a basic knowledge of the MIAPPE standard.
The exact listing of the metadata fields required for a FAIRification of the genotyping data set within a VCF file is also part of this recipe with examples and explanations (See details in Section 4.2).
FAIRification Objectives, Inputs and Outputs¶
Actions.Objectives.Tasks |
Input |
Output |
|---|---|---|
Table of Data Standards¶
Data Formats |
Terminologies |
Model |
|---|---|---|
Table of Software and Tools¶
Tool Name |
|---|
Step-by-Step Process¶
Step 1: Take DNA sample¶
The experimentalist takes a sample of plant biological material. The sample metadata are collected according to the MIAPPE specifications, Biological Material section. It enriches minimal MCPD fields with sample traceability information.
Step 2: Register sample material at BioSamples¶
This is done in general through JSON API (Python and Shell commands are also available). Refer to the official documentation for the complete details, for training material and slides regarding this, refer to 3. Here is the proposed procedure:
Create a user account
First you need an account to submit samples to EMBL-EBI BioSamples database. We recommend new users, or those planning to make downstream submissions to ENA, to use the Webin Authentication service. You can create a Webin account using Webin web interface. Please refer to ENA documentation for more details about creating an account.
Login to the system and get a JSON Web Token (JWT)
Webin uses JSON Web Token (JWT) for authentication. Use your login credentials to retrieve a JWT. You can either use the Webin Swagger interface (A) or a programmatic method (B):
A: Go to Webin Swagger and use POST /token endpoint to retrieve a JWT.
B: Use CURL or a programmatic method.
`TOKEN=$(curl -X POST "https://www.ebi.ac.uk/ena/submit/webin/auth/token" -H "accept: */*"
-H "Content-Type: application/json" -d "{\"authRealms\":[\"ENA\"],\"password\":\"PASSWORD\",\"username\":\"WEBIN_ID\"}")`
Allocate accessions via the pre-accessioning endpoint.
Use the pre-accessioning endpoint to reserve BioSamples ID. This will create private, empty samples with future release dates. The only mandatory field for pre-accessioning is the sample name.
The following CURL command returns 3 accessions as the body contains names for 3 samples.
`curl 'https://www.ebi.ac.uk/biosamples/samples/bulk-accession?authProvider=WEBIN'
-i -X POST -H "Content-Type: application/json;charset=UTF-8" -H "Accept: application/hal+json"
-H "Authorization: Bearer $TOKEN" -d '[{ "name" : "FakeSample 1"}, { "name" : "FakeSample 2"}, { "name" : "FakeSample 3"}]'`
Please refer to the BioSamples documentation.
More general information is available on the RDMkit 1. A specific checklist is used: BioSamples - Plant MIAPPE checklist.
Step 3: Perform sequencing of DNA sample¶
The sequencing staff performs the sequencing of the DNA sample, which is followed by a quality control. The reads are then archived in the institutional Laboratory Information Management System (LIMS).
Step 4: Register and submit sequencing reads to ENA¶
Submit Sequencing reads to ENA, using BioSamples IDs to identify material.
4.1 The Study¶
To begin, you should register a study. Recall that a study describes the purpose of the work you have done, groups other objects beneath it, and controls when the data becomes public. A study is required for all submissions to ENA.
Log in to the Webin Submission Portal with your Webin credentials.
Click the ‘New Submission’ tab and find the ‘Register study (project)’ radio button. Click ‘Next’ to see the study registration interface
EMBL-EBI Webin Submission Interface
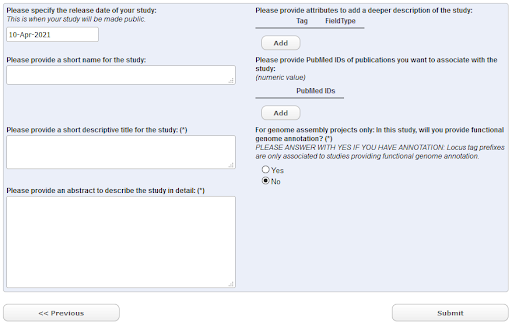
Fig. 27 EMBL-EBI Webin Submission Interface¶
The ‘Short Name’ field should be filled in with something brief and meaningful, e.g.:barley_study_2021
You should take time to provide a descriptive title and informative abstract for your own studies, but these can be edited later if needed. For now, use as your title: GBS Study of Barley from <Your Town/Lab>
When you have completed all required fields, click ‘Submit’ and then confirm.
Now navigate to the ‘Studies’ tab to see the study you just registered. You might need to refresh the page!
Make a note of its accession numbers (ERP#####, PRJEB#####), which will resemble. For a submission, these would be the numbers you would cite in any publications involving the data.
4.2 The Sample¶
The next step is to register the sample, which will give other users essential context for the sequence data you are submitting. The sample describes the source biological material of your sequencing work.
As discussed above, samples are best submitted through BioSamples.
In ENA, samples are required to conform to a checklist of values. Checklists define a set of mandatory and recommended descriptor fields for a given type of sample. It is recommended that you look at these early and make sure you collect all required metadata items for the type of sample you will be registering.
The selection of available checklists can be browsed at: https://www.ebi.ac.uk/ena/browser/checklists. Currently, three plant specific checklists are available: ERC000020, ERC000035 and ERC000037.
Return to the ‘New Submission’ tab in the submission service. You may need to click the ‘Restart Submission’ button at the bottom of the page.
Find the ‘Register samples’ radio button and click ‘Next’.
You must choose an appropriate checklist of values to be provided for your sample: click ‘Select checklist’ and expand the ‘Other checklists’ group to browse checklists of this type.
Select the appropriate checklist. For general purposes ERC000037 (‘ENA Plant Sample Checklist’) should be the default when submitting plant sequencing data. Now click ‘Next’. add an image of the web ui for https://www.ebi.ac.uk/ena/browser/view/ERC000037
Submitters now have the option of including additional fields in their checklist. It is not necessary to include any additional fields, but you can take this opportunity to see which fields are included by default, what requirements they have, and what else is available.
Click ‘Next’.
Now you have the opportunity to construct a template for a submission of many samples: notice the phrase ‘Template Basic Details’ at the top of the interface. This allows you to define values which are common to all samples in your submission.
Since we are only submitting one sample, skip this screen by clicking ‘Next’.
On the next screen, click the ‘+ Add’ button to create your sample.
Give a ‘Unique name’ to your sample:
barley_sample_1
Hint: This value must always be unique among all samples submitted by your account!
Give your sample a title, e.g.:
Barley sample from <Your Town/Lab> greenhouse
Write a brief description for your sample.
Find the appropriate taxon for your sample: For example enter ‘Hordeum vulgare’ into the box and select it when it appears.
Further taxonomic details are automatically filled below this box. This box will only accept values which match with a species-rank entry in the NCBI Taxonomy database.
Fill out the remaining details by reference to the values to the best of your knowledge. Only fields marked with a ***** are required, but it is best to fill as many fields as possible.
Use a markdown directive (note: not supported by hackMD but supported by jupyter notebook)
Note
You can always find out more about what a field means and what information it accepts by hovering over the blue ‘i’.
By the time you have completed this, your sample will be well-annotated and understandable to people finding it in the database later.
You are ready to submit when all the checks on the right of the page are green ticks, and none are red crosses.
Click ‘Submit’.
Now navigate to the ‘Samples’ tab to see the sample you just registered. You might need to refresh the page.
Make a note of its accession numbers as you will need these later:
ERS####### and SAMEA#######
4.3 The Read Data¶
Now that study and sample metadata have been registered, it is time to submit the read data you have produced.
Return to the ‘New Submission’ tab in the submission service. You may need to click the ‘Restart Submission’ button at the bottom of the page.
Find the ‘Submit sequence reads and experiments’ radio button and click
Next, you are asked to choose which study these reads should be added to. Select the study you registered earlier and click ‘Next’.
Now, you have the opportunity to register samples as part of this submission. However, you already registered a sample, so click ‘Skip’.
Now, select ‘One Fastq file (Single)’ from the list of file options.
Fill out the form to describe your experiments and register the files. Ordinarily, you would need to upload the files to a staging area before submitting them through this interface, refer to the ENA upload guidelines.
Appropriate metadata for your read data submission should look similar to this:
Sample reference:
Instrument Model: Illumina HiSeq 2500
Library Name: barley_library_1
Library Source: GENOMIC
Library Selection: Restriction Digest
Library Strategy: GBS
Library Layout: SINGLE
First File Name: HOR_1361_BRG.fastq.gz
Note
If you did not note down your sample accession, you can safely visit the ‘Samples’ tab, copy the accession, then return to the New Submission tab: your progress will be saved.
Click ‘Submit’ and see if your submission validates successfully. If you encounter errors, try using the ‘Download Template Spreadsheet’ button, open the file and check it in this plain text format; it can be easier to fix errors this way than in the interface.
Note
Note that the use of template spreadsheets is the best way to submit multiple datasets through this interface.
If you manage to complete your submission, visit the ‘Runs’ tab to review your submission.
Step 5: Check if used reference genome assembly is available¶
Is a GCF / GCA accession number available ? Check on https://www.ebi.ac.uk/ena/browser.
If yes, proceed directly to VCF submission at step 4.
If no, submit reference genome assembly to INSDC (NCBI Genbank / EMBL-EBI ENA / DDBJ) and wait until accession number is issued, then proceed to step 4.
Step 6: Analyse sequencing results¶
The bioinformatician performs the computational analysis, then the genotyping results are archived into the LIMS.
Step 7: Prepare genotyping dataset for submission of VCF file to EVA¶
In order to ensure interoperability of VCF files, in accordance with the good practice outlined in 2, the following VCF meta-information lines should be used:
Recommended meta-information lines :¶
##fileDate(Date): creation date of the VCF in the basic form without separator: YYYYMMDD
Example:
##fileDate=20120921
##bioinformatics_source (URL or URI): Analytic approach usually consisting of chains of bioinformatics tools for creating the VCF file specified as the DOI of a publication, or more generally as URL/URI, like a public repository for the scripts used. The preferred way to describe this would be to use WorkflowHub.eu (possibly in CWL format) and to be fully transparent about the bioinformatics toolchain used to generate the results.
Example:
##bioinformatics_source="doi.org/10.1038/s41588-018-0266-x"
##reference_ac (assembly_accession): accession number, including the version, of the reference sequence on which the variation data of the present VCF is based.
Example:
##reference_ac=GCA_902498975.1
##reference_url (DOI): a DOI (or URL/URI) for downloading of this reference genome, preferably from one INSDC archive.
Example:
##reference_url="ftp.ncbi.nlm.nih.gov/genomes/all/GCA/902/498/975/GCA_902498975.1_Morex_v2.0/GCA_902498975.1_Morex_v2.0_genomic.fna.gz"
##contig (<ID=ctg1, length=sequence_length, assembly=gca_accession, md5=md5_hash, species=NCBITaxon:id>) : The individual sequence(s) of the reference genome.
Example:
##contig=<ID=chr1H,length=522466905,assembly=GCA_902498975.1,md5=8d21a35cc68340ecf40e2a8dec9428fa,species=NCBITaxon:4513>
##SAMPLE(<ID=\(BioSample_accession, DOI=\)url, ext_ID=$registry:identifier>) : Describe the material whose variants are given in the genotype call columns in greater detail and can be extended using the specifications of the VCF format.
In case no DOI exists and the material is held by a FAO-WIEWS (https://www.fao.org/wiews/background/en/) recognised institution, the external ID consists of the FAO-WIEWS instcode, the genus and the accession number (see example 2). If the database is not registered with FAO-WIEWS and is not available under a DOI, the DNS of the holding institution, the database identifier, the identifier scheme and the identifier value should be provided (see example 3). For multiple external IDs the field should be used multiple times (delimited by commas).
Examples:
One genotype from a barley (Hordeum vulgare) GBS experiment with a DOI registered.
`##SAMPLE=<ID=SAMEA104646767,DOI="doi.org/10.25642/IPK/GBIS/7811152">`One genotype from a barley (Hordeum vulgare) GBS experiment with the FAO-WIEWS code available but no DOI.
`##SAMPLE=<ID=SAMEA104646767,ext_ID="DEU146:Hordeum:HOR 1361 BRG">`One genotype from a barley (Hordeum vulgare) GBS experiment with no DOI and no FAO-WIEWS code available.
##SAMPLE=<ID=SAMEA104646767,ext_ID="ipk-gatersleben.de:GBIS:akzessionId:7811152">
In case of adding new fields :¶
Please check the official format specifications to avoid redundancy and possible incompatibilities.
Step 8: Submit VCF file to EVA¶
Once the metadata and data has been formatted according to the specifications above, make sure that the resulting VCF file complies with VCF specifications. For that purpose, we propose the VCF validator tool on GitHub
vcf_validator -i /path/to/file.vcf
vcf_validator -i /path/to/compressed_file.vcf.gz
Once the file has been fully validated without any error messages, you can submit the VCF file to EVA, using BioSamples IDs to identify the material, GCF/GCA accession for the reference genome assembly, and ENA accession numbers for the sequencing reads of the material used. Refer to the official documentation.
Conclusion¶
At this point, the VCF contains metadata and data formatted for the purpose of better discoverability and higher interoperability. Data could thus be more easily read and evaluated automatically by machines, and it is made easier to connect different data sources with each other, so that in general a higher degree of FAIR has been achieved.
What to read next?¶
Learn more about:
FAIRsharing records appearing in this recipe:
- Common Workflow Language (CWL)
- DNA Data Bank of Japan (DDBJ)
- Digital Object Identifier (DOI)
- European Nucleotide Archive (ENA)
- European Variation Archive (EVA)
- FASTA Sequence Format (FASTA)
- FASTQ Sequence and Sequence Quality Format
- Fungal Gross Anatomy Ontology (FAO)
- JavaScript Object Notation (JSON)
- Minimum Information about Plant Phenotyping Experiment (MIAPPE)
- The FAIR Principles (FAIR)
- Uniform Resource Identifier (URI)
- Uniform Resource Locator (URL)
- Variant Call Format (VCF)
- WorkflowHub
Reference¶
References
- 1
A. F. Adam-Blondon, C. Pommier, D. Faria, E. Le Floch, P. Lieby, and S. Beier. Tool assembly - Plant Genomics. URL: https://rdmkit.elixir-europe.org/plant_genomics_assembly.html.
- 2
S. Beier, A. Fiebig, C. Pommier, I. Liyanage, M. Lange, P. J. Kersey, S. Weise, R. Finkers, B. Koylass, T. Cezard, M. Courtot, B. Contreras-Moreira, G. Naamati, S. Dyer, and U. Scholz. Recommendations for the formatting of Variant Call Format (VCF) files to make plant genotyping data FAIR [version 1; peer review: 1 approved with reservations]. F1000Res, 2022. doi:10.12688/f1000research.109080.1.
- 3
M. Brouwer, C. Pommier, A. Fiebig, S. Beier, M. van Kaauwen, I. Liyanage, S. Holt, F. Xu, and B. Koylass. ELIXIR FONDUE Datathon. URL: https://github.com/PBR/elixir-fondue-datathon.
Authors¶
Authors
Name |
ORCID |
Affiliation |
Type |
ELIXIR Node |
Contribution |
|---|---|---|---|---|---|
Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) |
|
Writing - Original Draft |
|||
Université Paris-Saclay, INRAE, BioinfOmics |
|
Writing |
|||
Université Paris-Saclay, INRAE, BioinfOmics |
|
Writing |
|||
Isuru Liyanage |
EMBL-EBI |
|
Writing |
||
EMBL-EBI |
|
Review & Editing |
|||
University of Oxford |
|
Review & Editing |